En la década de 1950 se descubrió que los genes no eran nada más y nada menos que cadenas de nucleótidos. Un descubrimiento que parece tan sencillo desencadenó, sin embargo, el nacimiento de una nueva era: la de la tecnología del ADN recombinante.
La primera molécula de ADN recombinante fue creada por Paul Berg, a comienzos de los 70. Para aquella época, los biólogos moleculares habían aprendido a alterar genes individuales, cortar y pegar pedazos de ADN de diferentes organismos y moverlos de uno a otro.
Otros pioneros de esta tecnología fueron Stanley Cohen, un genetista estadounidense, y Herbert Boyer, un bioquímico de la misma nacionalidad . Cohen estaba interesado en aprender cómo los genes de plásmidos otorgan resistencia a las bacterias y Boyer trabajaba con enzimas de restricción (ver apartado 3.3.), por lo que decidieron trabajar juntos en la combinación de dos plásmidos resistentes a antibióticos diferentes para crear una bacteria resistente a ambos.
ADN recombinante es una molécula que proviene de la unión artificial de dos fragmentos de ADN. Por lo tanto, la tecnología de ADN recombinante es el conjunto de técnicas que permiten aislar un gen de un organismo, para su posterior manipulación e inserción en otro diferente. De esta manera podemos hacer que un organismo (animal, vegetal, bacteria, hongo) o un virus produzca una proteína que le sea totalmente extraña.
Estas técnicas se emplean normalmente para la producción de proteínas en gran escala, ya que podemos hacer que una bacteria produzca una proteína humana y lograr una superproducción, como en el caso de la insulina humana, que actualmente es producida por bacterias en grandes recipientes de cultivo, denominados biorreactores. Como las bacterias se multiplican muy rápidamente y pueden expresar grandes cantidades de proteínas, es posible lograr una sobreproducción de la proteína deseada. A esto justamente se dedica la biotecnología, es decir a la utilización de organismos vivos o de sus productos con fines prácticos.
El desarrollo de la tecnología del ADN recombinante fue posible gracias a varias líneas de investigación: 1) el conocimiento de las enzimas de restricción, 2) la replicación y reparación de ADN, 3) la replicación de virus y plásmidos y 4) la síntesis química de secuencias de nucleótidos.
En 1975 Daniel Nathans y Hamilton O. Smith descubrieron un tipo de proteínas -las enzimas endonucleasas o enzimas de restricción- que actúan como "tijeras moleculares", cortando la doble cadena de ADN a través del esqueleto de fosfatos sin dañar las bases. El descubrimiento de estas enzimas condujo a dichos microbiólogos al Nobel en 1978 y dio origen a la ingeniería genética.
Las enzimas de restricción son producidas por bacterias como método de defensa contra virus y degradan el ADN extraño.
A su vez, el propio genoma bacteriano está protegido contra sus enzimas de restricción mediante metilaciones (es decir, el agregado de un grupo metilo [-CH3)]) en un átomo específico de ciertos nucleótidos.
Estas moléculas son indispensables para la ingeniería genética, ya que producen fragmentos que se pueden unir entre sí fácilmente (con la ayuda de un "pegamento molecular": la enzima ligasa).
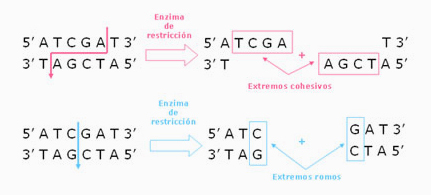
Las enzimas de restricción cortan dejando extremos cohesivos o romos. Los extremos cohesivos son generados cuando la enzima corta las dos hebras asimétricamente, dejando los extremos de cada hebra de simple cadena complementarios entre sí. Por otro lado, los extremos romos son generados cuando la enzima corta las dos hebras por el mismo lugar, generando dos extremos doble cadena.
Figura 1. Enzimas de restricción. Este tipo de endonucleasas puede dejar dos tipos de extremos. En el primer caso, el corte genera nucléotidos de simple cadena llamados extremos cohesivos. Estos extremos se pueden unir por medio de otra enzima, la ADN ligasa. En el segundo caso, se generan extremos doble cadena (extremos romos). Estos extremos también pueden ser unidos con la ayuda de una enzima ligasa pero, como no los extremos no son complementarios, la unión será más inespecífica.
Las enzimas de restricción permiten cortar el genoma de cualquier organismo en pequeños fragmentos llamados fragmentos de restricción. La colección de miles o millones de estos fragmentos se llama biblioteca génica.
Una vez que los biólogos encontraron cómo fabricar ADN recombinante usando enzimas de restricción y ligasas, el desafío siguiente fue cómo producir grandes cantidades de genes y cómo introducirlos en bacterias u otras células huésped. El primer problema fue solucionado con el uso de plásmidos, pequeñas moléculas de ADN circular presente en muchas bacterias.
Los plásmidos contienen uno o más genes de resistencia a antibióticos y son capaces de autorreplicarse, ya que contienen una secuencia de iniciación. Esto les permite replicarse de manera independiente del ADN genómico.
Podemos crear una molécula de ADN recombinante usando un plásmido. Este proceso consta de las siguientes etapas:
Esta es una manera relativamente eficaz de obtener millones de copias del ADN incorporado. Dado que todas las copias del gen provienen de una sola molécula multiplicada a partir de una única bacteria que dio origen a la colonia, esta técnica lleva el nombre de clonación. El término clon proviene de la jardinería; desde hace siglos los jardineros generan plantas nuevas a partir de gajos. Estas plantas son genéticamente idénticas y constituyen un clon.
Un clon es un grupo de células u organismos genéticamente idénticas.
El uso fragmentos de ADN como vectores cumple un rol fundamental en la ingeniería genética, ya que sirven para transferir material genético de un organismo a otro.
Vector: cualquier organismo o virus capaz de mover genes de un organismo a otro
Durante las décadas de 1970-1980, la manera más práctica de hacer múltiples copias de una secuencia particular de ADN era introduciendo una molécula de ADN recombinante (un plásmido más un gen) en una célula huésped. Esto podía resultar un poco engorroso, ya que muchas veces se disponía de una cantidad muy pequeña de moléculas de ADN para realizar pruebas.
A mediados de la década de los 80, la invención de una técnica capaz de generar centenares de miles de copias de una secuencia sin la necesidad de clonarla en ningún tipo de vector resolvió este problema. Kary Mullis, un bioquímico americano que trabajaba sintetizando ADN, resolvió este problema con la invención de la reacción en cadena de la polimerasa.
Esta se basa en la separación de ambas hebras de ADN, la posterior unión de pequeños fragmentos (denominados primers) y la extensión de estos fragmentos usando de molde el ADN de la muestra. Así se obtienen dos copias idénticas por cada molécula en cada ciclo de reacción. Para llevar a cabo esta reacción se requiere de una enzima polimerasa que sea capaz de soportar la alta temperatura del proceso de desnaturalización. Esta enzima es una ADN-polimerasa especial obtenida de una bacteria termófila (la Thermus aquaticus), resistente a altas temperaturas.
La reacción en cadena de la polimerasa permite amplificar la muestra de manera espectacular y su sensibilidad es tal que alcanza con una molécula para que la reacción genere una infinidad de copias. Esto la convierte en una importante, si no imprescindible, herramienta para el análisis de filiación o de criminalística forense, ya que con sólo una pequeñísima muestra se pueden realizar diferentes estudios comparativos que nos permiten conocer el dueño de un "rastro" genético en particular.
También se usa para el desarrollo de nuevas estrategias de diagnóstico médico, como la detección de virus o de mutaciones que provocan enfermedades genéticas, a partir de una muestra de ADN tan pequeña como un cabello o una gota de sangre.
Otra aplicación de la técnica de la reacción en cadena de la polimerasa es la transcripción inversa de ARNm. Mediante el uso de una enzima de origen viral denominada transcriptasa reversa puede usarse como molde una molécula de ARNm, transcribirla a una secuencia de ADN, y luego ser amplificada como cualquier otra secuencia por la reacción en cadena de la polimerasa, con lo que se obtiene una gran cantidad de ADN a partir de unas pocas moléculas de ARNm. Esta técnica es muy empleada para el estudio de expresión génica, así como para la producción de proteínas en diferentes organismos.
Hasta hace relativamente poco, encontrar un gen en un genoma era como encontrar una aguja en un pajar. Si tuviéramos que realizar dicha tarea, probablemente usaríamos un imán para atraer la aguja (siempre y cuando esta sea de metal y posea propiedades magnéticas). De manera análoga, hoy día podemos encontrar genes o proteínas usando, respectivamente, sondas y anticuerpos, que actúan como imanes moleculares.
Las sondas son fragmentos de ADN o ARN de simple cadena complementarias a la región de ADN o ARN que queremos encontrar. Contienen alguna "marca" que permite revelar su unión (hibridación) a la secuencia elegida y de esa manera revela presencia de la región buscada.
¿Cómo se marcan esas sondas o fragmentos de ácidos nucleicos? Las sondas pueden ser "coloreadas" durante su síntesis utilizando nucleótidos marcados con isótopos radiactivos. Otra manera de marcarlas es mediante la unión con una molécula que pueda ser detectada posteriormente como fluorescente, un marcador químico que puede ser detectado por un anticuerpo, o una enzima cuya actividad produce el cambio de color de su sustrato.
Una de las técnicas más empleadas para localizar fragmentos determinados del genoma (o la expresión de algún gen en particular por medio de la detección de la presencia de su correspondiente ARNm) es la hibridación in situ. Durante este proceso se incuba el ADN con una sonda (de ADN o ARN) complementaria a la secuencia buscada, y marcada radiactiva o químicamente.
La sonda "navega" por el interior de la célula hasta encontrar una secuencia complementaria a ella. Una vez que esto ocurra se formará una doble cadena que permitirá no sólo detectar la presencia de esta secuencia, sino indicar el lugar específico que ocupa dentro de la célula. Si se tratara de un fragmento de un cromosoma, nos permitiría saber su localización exacta o locus.
Para revelar la presencia de hibridación de la sonda, la muestra debe exponerse a una placa radiográfica; en el caso de las sondas marcadas químicamente se procede a su revelado mediante el uso de moléculas fluorescentes o que puedan ser detectadas por colorimetría (utilizando una enzima que provoque el cambio de color de un sustrato).
Se puede también utilizar colonias de bacterias con ADN recombinante con una técnica similar, pero que usa anticuerpos en vez de sondas. Se comienza por una colonia de bacterias que contengan y expresen el gen de ADNc. Luego se trata las bacterias con un anticuerpo marcado, que se unirá a la colonia que sintetice la proteína deseada. De esta manera se puede localizar el gen en cuestión y clonarlo para obtener múltiples copias.
Si visitamos un laboratorio de biología molecular, probablemente encontraremos una cuba con un gel al cual se le aplica una corriente eléctrica. Este método, llamado electroforesis en gel, es muy usado para separar moléculas de diversos tamaños y es la base de las técnicas que describiremos para identificar ADN, ARN, y proteínas. En los párrafos siguientes describiremos entonces la electroforesis para poder comprender luego cómo funcionan las demás técnicas.
La electroforesis en gel es uno de los métodos más utilizados en los laboratorios para separar ácidos nucleicos o proteínas, de acuerdo con su tamaño. Esto se logra por la diferencia de desplazamiento de las moléculas a lo largo de un gel sometido a un campo eléctrico. La carga eléctrica sirve como fuerza impulsora que atrae las moléculas cargadas negativamente hacia el otro extremo del gel que posee carga positiva. Existen distintos tipos de gel; comúnmente se utiliza agarosa o poliacrilamida.
La velocidad de desplazamiento de las moléculas es inversamente proporcional a su tamaño. En los ácidos nucleicos la carga está dada por los nucleótidos, por lo que el tiempo que tardará la molécula en atravesar el gel será directamente proporcional al número de nucleótidos que tenga, es decir a su tamaño. De esta manera, para un tiempo determinado, las moléculas más grandes quedarán más retrasadas en el gel que las moléculas más pequeñas.
La separación de proteínas es un poco mas complicada, ya que éstas poseen una estructura tridimensional compleja y compacta, además de una carga neta que depende de la identidad de los aminoácidos que la componen. Esto hace más difícil separarlas sólo por su tamaño y por eso se emplea una versión modificada conocida como electroforesis en gel de poliacrilamida. Las proteínas forman bandas de acuerdo con su tamaño; si se corre una muestra con proteínas de tamaño conocido se puede determinar el tamaño de la proteína estudiada por la distancia recorrida en el gel. Las proteínas más grandes quedarán arriba y las más chicas quedan abajo (y las de tamaño medio quedarán entre las demás). Además, el grosor de la banda obtenida dependerá de la cantidad de proteína que se haya sembrado; cuanto mayor sea la cantidad de proteínas, más gruesa será la banda.
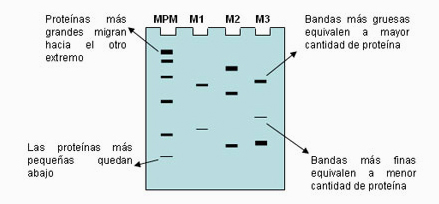
Figura 1. Electroforesis en gel. Se siembra la muestra (ADN, ARN o proteínas) en un gel que es sometido a una corriente eléctrica. Las moléculas se mueven a través del gel impulsadas por dicha corriente. Esta técnica permite separar moléculas de acuerdo a su tamaño y carga ya que las más pequeñas (o aquellas con mayor carga) se mueven con mayor velocidad. El gráfico muestra bandas de distinto ancho, correspondientes a distinta cantidad y tamaño de la muestra de proteínas.
Existen diferentes técnicas que emplean la separación en gel por electroforesis como primer paso para identificar la presencia de determinadas moléculas dentro de una muestra. Una de ellas fue inventada por Edward M. Southern en 1975 para la detección de ADN. Una vez finalizada la corrida en el gel, se realiza la transferencia (o blotting) de las muestras a una membrana para su posterior revelado. Luego se utilizó el mismo sistema para detectar ARN y se lo denominó Northern Blot (en clara alusión al nombre del científico inventor de la técnica basada en ADN).
Estas dos técnicas consisten en una electroforesis seguida de transferencia y emplean secuencias específicas (sondas) complementarias a la molécula de ácido nucleico a identificar. Es decir, ambas utilizan la hibridación de ácidos nucleicos para la detección de las moléculas buscadas. Los principios generales son los mismos, pero hay diferencias en el procedimiento debidas principalmente a que el ARN se degrada mucho más fácilmente que el ADN; esto hace necesario tomar muchas más precauciones.
Una vez sembradas las muestras en un gel, se realiza una corrida electroforética hasta lograr que las moléculas se separen por tamaños de manera tal que se puedan distinguir las buscadas. Luego se transfieren los ácidos nucleicos del gel a la membrana. De esta manera los que quedan retenidos pueden ser sometidos a ensayos de hibridación con sondas específicas para detectar la presencia de determinadas secuencias.
Estas dos técnicas persiguen fines distintos: el Southern Blot, que detecta la presencia de ciertas secuencias en el ADN, se usa, por ejemplo, para estudiar mutaciones en el genoma. El Northern, en cambio, permite detectar qué genes son transcriptos en determinadas condiciones. Hemos visto que dos células del mismo organismo se distinguen por los genes que expresan y no por su ADN. El Southern Blot nos permite evidenciar el mismo material genético y el Northern Blot, la expresión de diferentes genes.
Si luego queremos detectar proteínas, usaremos otra técnica similar: el Western Blot, que deriva de la electroforesis en gel y separa proteínas mediante la utilización de anticuerpos específicos. Una vez que la muestra con las proteínas que se quiere identificar ha corrido en un gel, se la transfiere a una membrana especial donde las proteínas quedan "ancladas" o adheridas. Después, se incuba esta membrana con el anticuerpo específico contra la proteína blanco, y se revela. Con esta técnica pueden obtenerse datos aproximados del tamaño de la proteína y su concentración en la muestra sembrada.
Otra técnica que emplea la hibridación de sondas específicas marcadas es la de microarreglos (Microarrays), que consta de una membrana dividida en celdillas que poseen adheridas sondas que representan un gen en particular. En conjunto, estas celdillas contienen todos los genes conocidos de un organismo. De esta manera, cuando se coloca una muestra en estos chips se irá hibridando cada gen que se encuentre en la muestra con su sonda correspondiente en la casilla determinada. Cuando una celdilla da positivo, quiere decir que ese gen determinado está presente en la muestra.
Los ensayos con microarreglos son rápidos de realizar, pero muy engorrosos de analizar. Además, están muy limitados a los genes que ya se conocen. Permiten obtener un pantallazo rápido sobre qué está pasando en la célula en ese momento determinado, en cuanto a qué genes están encendidos y cuáles apagados. Sin embargo, los resultados obtenidos nunca deberían ser concluyentes, sino que tendrían que ser validados mediante otras técnicas (por ejemplo, Northern Blot).

Microarreglos: esta técnica permite analizar simultáneamente miles de genes.